Server For Computing/Predicting DEPTH, Ligand Binding Sites, pKa, Disulfide engineering
Server For Computing/Predicting DEPTH, Ligand Binding Sites, pKa, Disulfide engineering
Users can specify the 4-letter code of an existing protein structure in the PDB
Example: 2FP7, 2grn
Users also have the option to upload a file in PDB format.
In its current implementation, the server only processes 5 structures per submission. For
larger scale applications, the program can be downloaded and run locally.
For Membrane proteins, the structures are used from local copy of
OPM database.
You can upload your own structure with membrane calculated by
PPM server.
A residue-wise plot of depth value is displayed. The plot shows both mean and standard deviation of depth values. The depth output is also available for download in tab-delimited and PDB formats. In the PDB format depth values are recorded in the b-factor column.
Using an extensive benchmark of 900 ligand bound proteins, we have established that some residues in most ligand binding cavities are simultaneously surface exposed and deep.
The server predicts small molecule binding cavities on proteins. Prediction results may vary
with choice of depth computation parameters. For example, using a larger value of minimum number
of neighborhood waters results in the detection of larger cavities, which maybe apt for larger
ligands.
The algorithm estimates the probability value of forming part of a binding cavity for every
residue of the protein. Users have the option to alter the recommended cavity
prediction probability threshold.
Binding cavity prediction for other biomolecule other than proteins (DNA, RNA etc) is currently not supported. (Please contact author for this application)
The algorithm estimates the probability value of forming part of a binding cavity for every residue of the protein. A residue-wise probability plot is displayed. A list of residues that have probabilities greater than the threshold are displayed along with the list of residues predicted to form the binding cavities. Users can also choose to download the results in PDB (with b-factor column replaced by probability value) or tab-delimited formats.
Normalized residue-wise solvent accessible surface area of protein residues are computed using the Shrake-Rupley algorithm [Shrake and Rupley, 1973]. The output is downloadable in tab-delimited format. The surface area is calculated for 5 categories of atoms including - all atoms, Main Chain, Side Chain, Polar Side Chain and Non-Polar Side Chain (i.e. carbon atoms).
For multi domain protein, the recommended values are
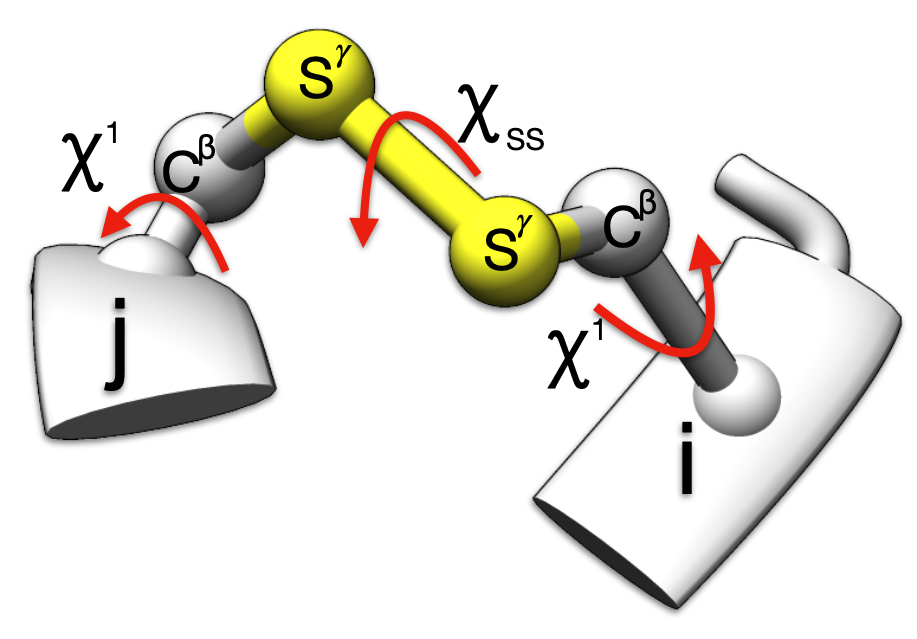
1.6 ≤ rss ≤ 2.4 and 60° ≤ |𝜒ss| ≤ 120°
and
30° ≤ |𝜒i1| ≤ 90° or 150° ≤ |𝜒i1| ≤ 180°
and
30° ≤ |𝜒j1| ≤ 90° or 150° ≤ |𝜒j1| ≤ 180°
1.6 ≤ rss ≤ 2.4 and 60° ≤ |𝜒ss| ≤ 120°
and
30° ≰ |𝜒i1| ≰ 90° or 150° ≰ |𝜒i1| ≰ 180°
and
30° ≰ |𝜒j1| ≰ 90° or 150° ≰ |𝜒j1| ≰ 180°
1.6 ≰ rss ≰ 2.4 or 60° ≰ |𝜒ss| ≤ 120°
Si or Sj cannot be geometrically fixed