Users can submit either a sequence or a structure as input. For input sequences, homology models would be built
for depth calculation. Input structures (structures from the PDB or user uploaded models) would be used without
checking for model accuracy.
For both sequence and structure inputs, several submission options are supported.
Users can submit a plain text of protein sequence through textbox provided.
For
multiple sequences, a fasta format should be used (separate sequences by a line starting
with '>'). Protein 3D model and prediction is made for each sequence separately.
User can submit a sequence by its GI / Accession number.
The server will fetch the
corresponding sequence from
NCBI for the prediction.
User can submit a sequence by uploading a file with sequence(s) in FASTA format.
User can submit a protein structure by its 4-letter PDB code, if the structure is
deposited in Protein
Data Bank .
The server will fetch the structure from PDB server for the
prediction.
User can submit a protein structure by uploading a file in PDB format.
For a submitted job, the server will return an url and a job id for user to retrieve the output.
Upon job completion, the page will refresh to show the output.
If specified, users would be intimated of the results via e-mail (sample screenshot).
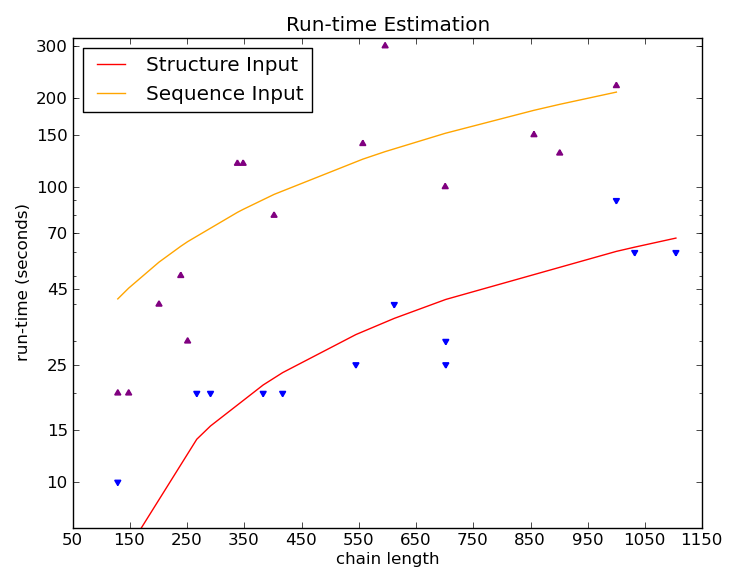
The estimated server running time for submitted temperature sensitive mutant prediction jobs. The running times for sequence and structure inputs were estimated using 14 protein sequences (chain lengths ranging from 129 - 1000 amino acids) and 13 protein structures ( chain lengths ranging from 129 - 1104 amino acids) respectively. The data were fitted to logarithmic curves.
(click here to see details for running time estimation)
note: Actual run times may also depend on how long the job spends in queue. Users are advised to solicit for results to be sent via e-mail.
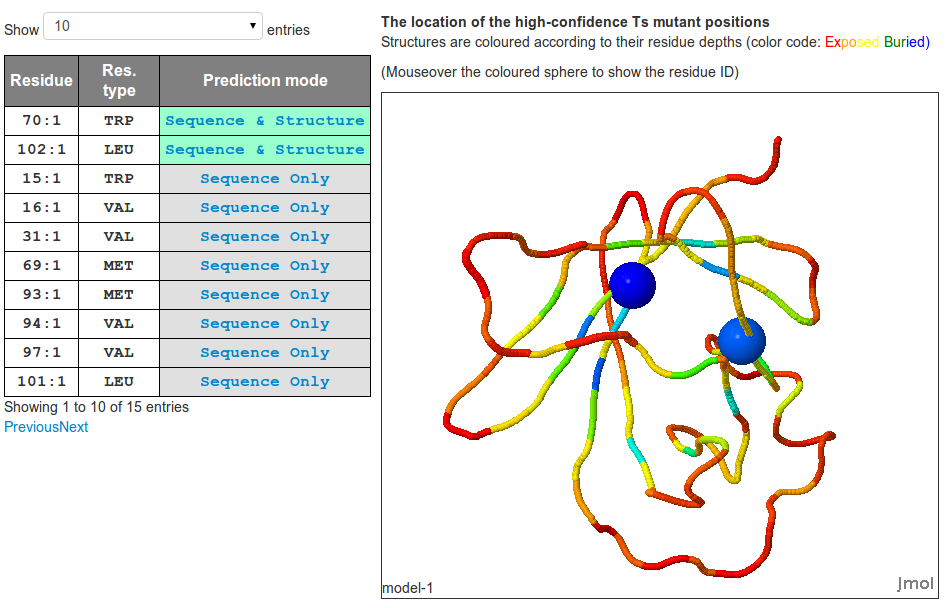
The prediction result will be summarized in a table. Predicted Ts mutant positions, residue type and their
prediction modes are listed. We recommend mutating high confidence predictions by both sequence- and structure-
based method first before examining predictions of lower confidence level (i.e. predictions made by only
sequence-based only or only structure-based method only).
If a homology model is built, its 3D model will be rendered in a Jmol plugin.
Users can also examine target-template sequence alignment used in the homology
modeling (if performed), toggle
between simplified and advanced table (containing additional information used for predictions) and download
all results in a zipped file.
Results of TSpred are available for download (as a zipped file). Depending on your input method, all or several of the following files can be found in the unzipped directory:
*.pdb
Input protein structure file in PDB format
*.depth.pdb
PDB file with b-factor column replaced with residue/atom depth
*.fasta
Input sequence file in FASTA format
*.blast
PSI-BLAST result
*.atom.depth
atomic depth output
*residue.depth
residue depth output
predbur.out1
sequence-based prediction output
template_target.pap
template-target sequence alignment file in PAP format
TS_output.html
output rendered in html (i.e. the server output page)
Tspred_output.tsv
output in tab-separated format